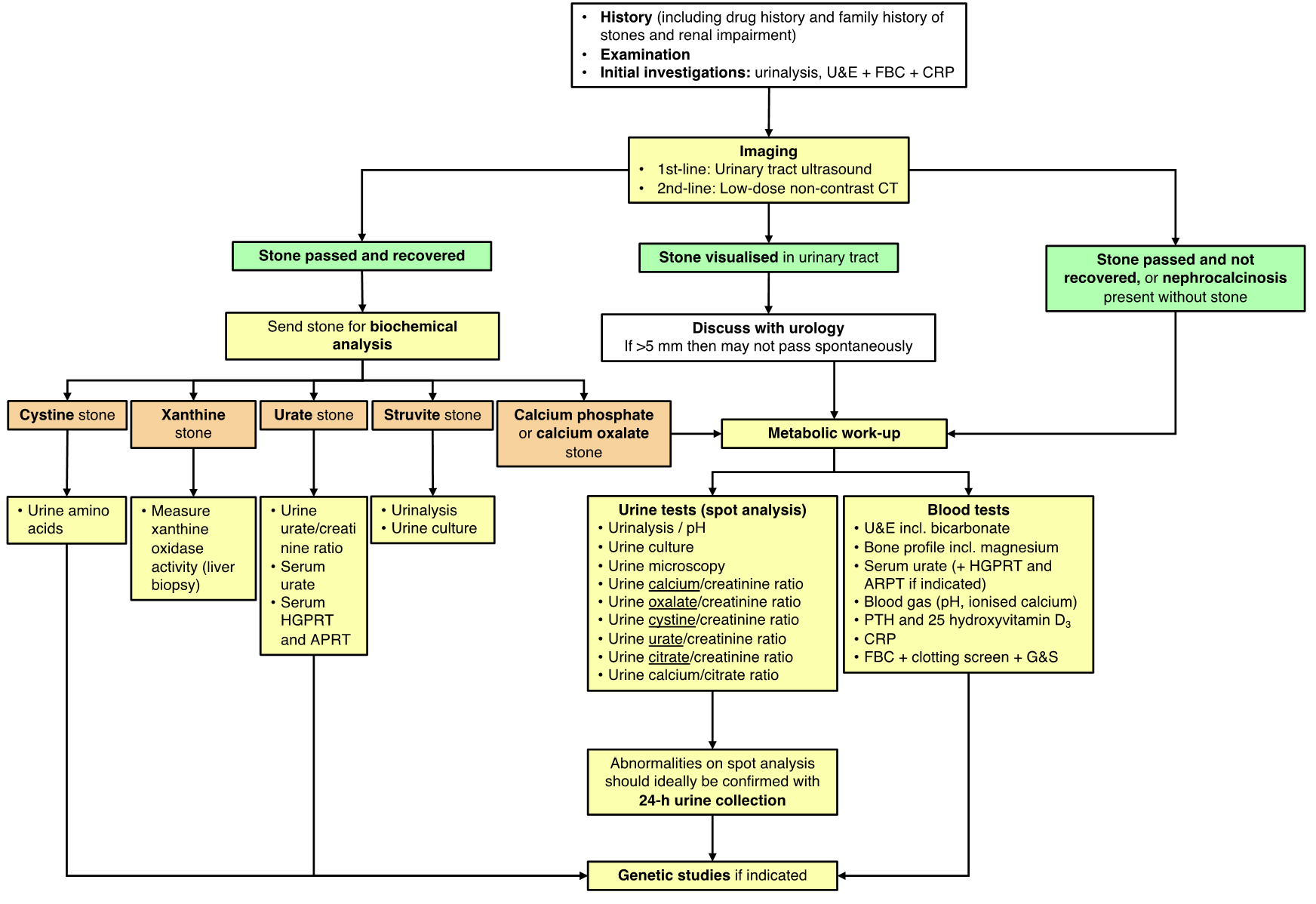
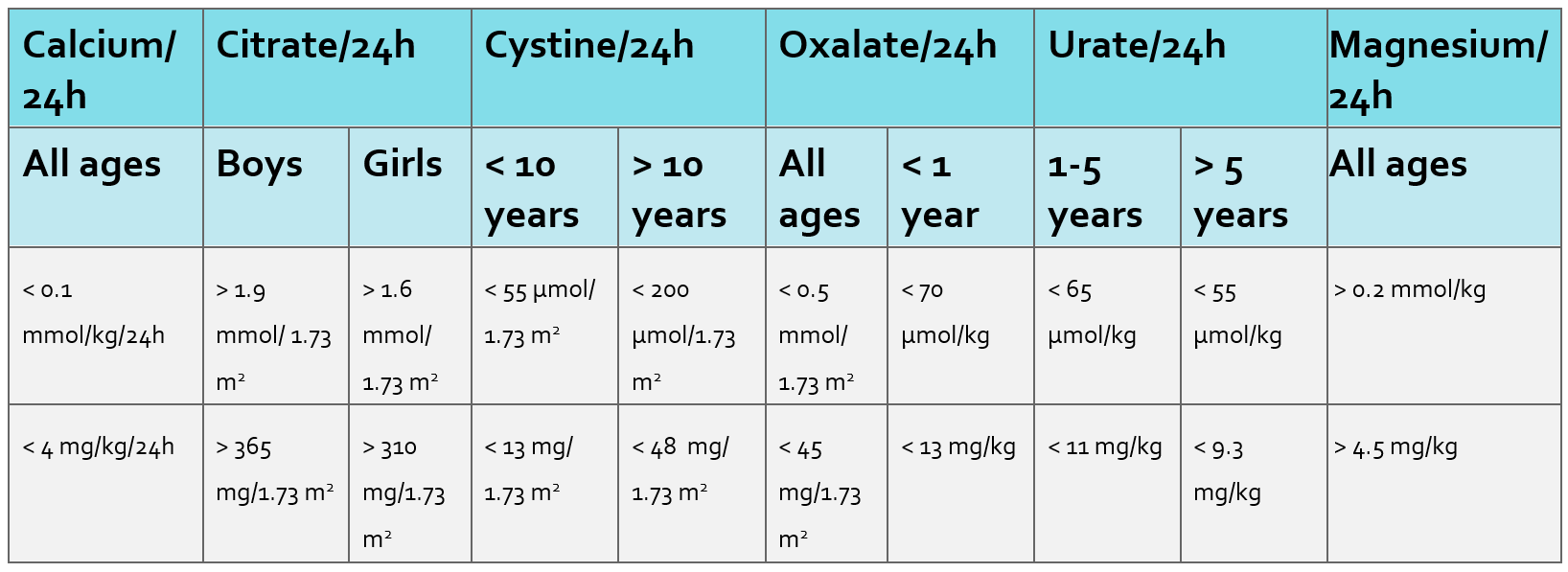
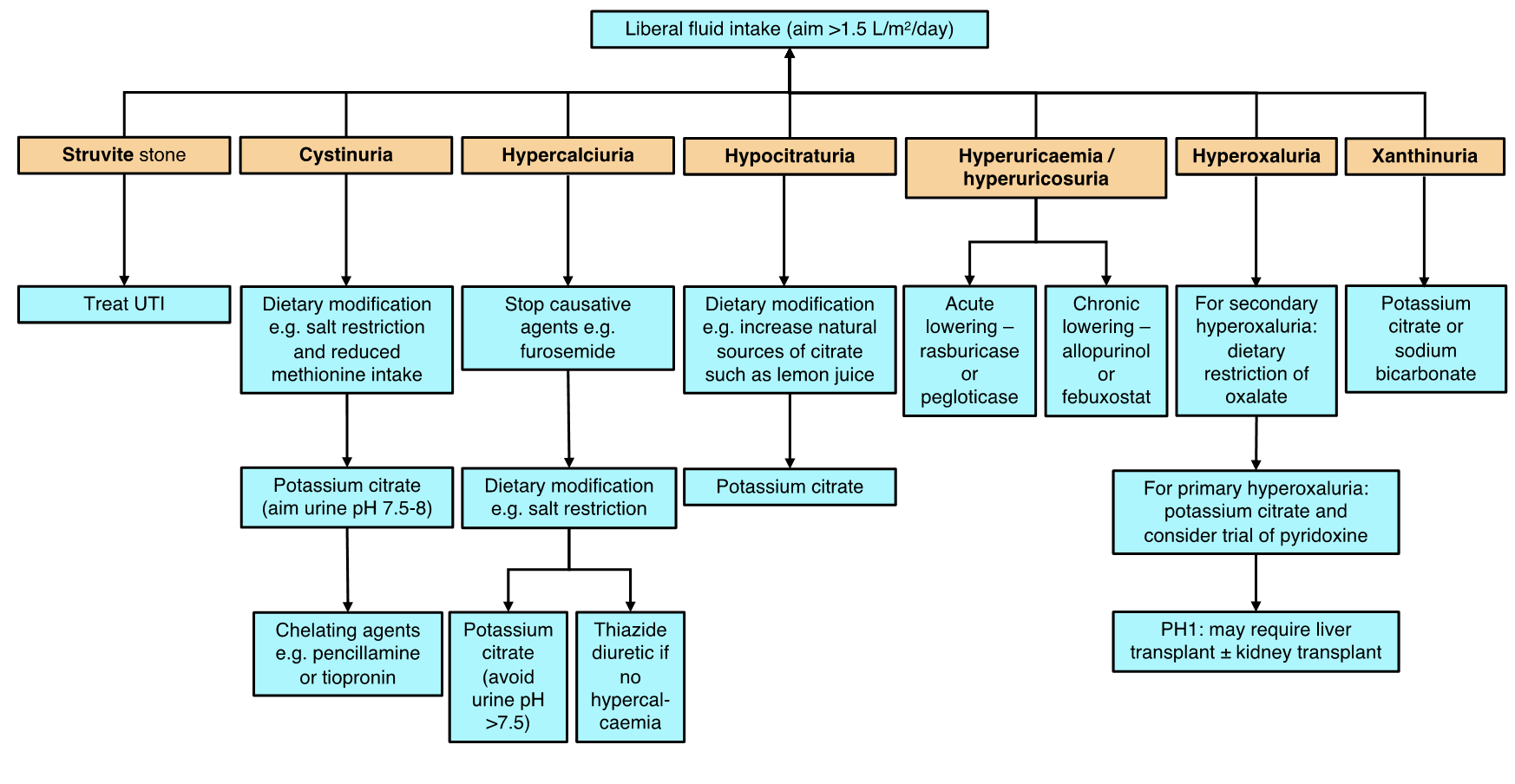
There is considerable overlap in the causes of NC and UL. NC is a risk factor for urolithiasis (UL) but does not necessarily lead to the development of stones.
- Hypercalciuria (which may occur in the context of hypercalcaemia or normocalcaemia):
- Idiopathic hypercalciuria – the most common cause of hypercalciuria
- Increased sodium/salt intake (leads to increased distal tubular secretion of calcium)
- Vitamin A, C and D excess or intoxication
- Prolonged immobility due to increased bone resorption leading to resorptive hypercalciuria
- Hyperparathyroidism – may be primary, secondary or tertiary
- Hypophosphatasia
- Hypophosphataemia
- Ketogenic diet
- Malignancy with paraneoplastic effects
- Sarcoidosis and other granulomatous diseases
- Milk-alkali syndrome – hypercalcaemia and metabolic alkalosis secondary to high intake of calcium (usually dietary supplements) and absorbable alkali (often in the form of antacid medications)
- Inherited disorders affecting the renal tubules (see below)
- Medications:
- Loop diuretics (e.g. furosemide) and some potassium-sparing diuretics (e.g. triameterene)
- Glucocorticoids (e.g. dexamethasone)
- Acetazolamide and other carbonic anhydrase inhibitors
- Topiramate
- Calcium supplementation
- Phosphate-containing laxatives (when taken in significant quantity such as in laxative abuse)
- Magnesium trisilicate
- Vitamins A, C and D (e.g. intoxication from parenteral nutrition)
- Ciprofloxacin
- Ceftriaxone
- Aciclovir
- Sulfa-containing medications
- Ethylene glycol – directly converted to glycolate and oxalate
- Protease inhibitors (e.g. indinavir and lopinavir)
- Probenecid
- Prematurity (particularly related to furosemide exposure)
- Inherited disorders:
- Distal / type 1 renal tubular acidosis (RTA) – a group of disorders characterised by impaired tubular H+ excretion leading to metabolic acidosis, elevated urine pH (> 5.5), hypocitraturia, hypercalciuria, hypokalaemia, and failure to thrive. Patients may also have sensorineural hearing loss.
- Bartter syndrome – a group of disorders characterised by impaired sodium, chloride and potassium reabsorption in the thick ascending limb of the loop of Henle. This leads to reduced calcium reabsorption and subsequent hypercalciuria. The biochemical disturbance is similar to that seen with loop diuretic therapy. The more severe antenatal/neonatal form is caused by variants in the SLC12A1 gene affecting the Na-K-Cl co-transporter.
- Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC) – autosomal recessive condition associated with impaired calcium and magnesium reabsorption in the thick ascending limb of the loop of Henle. Variants occur in the CLDN16 or CLDN19 genes encoding the tight junction proteins claudin 16 and 19. Patients with variants in CLDN19 also present with ocular abnormalities including myopia and nystagmus.
- Dent’s disease – a group of X-linked recessive conditions. The majority of cases are secondary to variants in the CLCN5 gene, which encodes a proximal tubular chloride channel. This leads to low molecular weight proteinuria and hypercalciuria, with the development of NC and progressive deterioration in kidney function over time.
- Cystinosis – an autosomal recessive lysosomal storage disorder due to variants in the CTNS gene encoding the protein cystinosin. This is associated with accumulation of the amino acid cystine. Patients develop proximal / type 2 RTA (renal Fanconi syndrome), hypercalciuria/NC, progressive renal impairment, growth failure and eye problems due to systemic cystine deposition.
- Lowe’s syndrome (oculocerebrorenal syndrome) – an X-linked recessive disorder due to variants in the OCRL1 The phenotype includes cataracts, hypotonia, developmental delay and type 2 RTA with hypercalciuria/NC.
- Wilson’s disease – an autosomal recessive disorder associated with impaired copper excretion. It may lead to type 2 RTA with associated hypercalciuria/NC.
- Tyrosinaemia – a group of autosomal recessive disorders associated with inability to break down the amino acid tyrosine. Patients may also develop a type 2 RTA with hypercalciuria/NC.
- Enamel-renal syndrome – autosomal recessive disorder due to variants in the FAM20A Patients have hypocalcaemia but present with NC.
- William’s syndrome – caused by chromosome 7 microdeletion. This predisposes to hypercalcaemia (cause not completely understood) and therefore hypercalciuria/NC.
- Hereditary hypophosphataemic rickets with hypercalciuria (HHRH) – an autosomal recessive condition due to variants in the NPT2C gene encoding the sodium-phosphate co-transporter. This results in urinary phosphate wasting, hypophosphataemic rickets, and inappropriately elevated 1,25(OH)2D (calcitriol) levels leading to hypercalciuria/NC.
- CYP24A1 gene variants – this gene encodes a member of the cytochrome P450 superfamily of enzymes. The protein product (CYP24A1) helps catalyse the degradation of 1,25(OH)2 Variants in CYP24A1 may lead to hypervitaminosis D and associated hypercalciuria/NC (particularly infantile hypercalcaemia/hypercalciuria).
- Sickle cell disease
- Metabolic disorders and abnormalities:
- Hyperoxaluria:
- Primary hyperoxaluria (PH types 1, 2 or 3) – a group of rare autosomal recessive conditions associated with excess oxalate production and elevated urine oxalate levels. PH type 1 (PH1) is the most severe phenotypic form. It has an estimated incidence of 1:100,000 and is caused by variants in the AGXT Patients typically present in childhood with NC, recurrent UL, and progressive renal failure. Serum oxalate levels typically remain normal (i.e. < 10 µmol/L) until they reach stage 4 chronic kidney disease (CKD). In PH1, other organs may also be involved including the bones (pain and anaemia), eyes (retinopathy), nerves (neuropathy) and heart (arrhythmia and heart block). PH2 is rarer still and patients may present around adolescence with oxalate stones. CKD develops in ~10% of PH2 patients. PH3 has the mildest phenotype. For all patient with PH, dietary modification is not helpful as oxalate production is a generated by metabolic pathways rather than from an external source.
- Secondary (enteric) hyperoxaluria – due to increased enteric oxalate absorption. This is associated with malabsorptive states such as short gut syndrome, inflammatory bowel disease and cystic fibrosis. Fat malabsorption causes increased binding of free fatty acids to dietary calcium. This thereby reduces the amount of calcium in the gut which would normally help to precipitate dietary oxalate. Furthermore, increased delivery of unabsorbed fatty acids and bile salts to the large intestine increases colonic permeability and leads to hyper-absorption of oxalate thus raising serum oxalate levels and predisposing to the formation of oxalate-containing stones.
- Idiopathic hyperoxaluria - high dietary consumption of oxalate (as seen in vegetarians) or low calcium intake may be predisposing factors to stone formation.
- Cystinuria – an inherited disorder with an estimated incidence of 1:10,000. It is an autosomal recessive condition caused by variants in the SLC3A1 or SLC7A9 genes respectively. It is associated with impaired tubular reabsorption of the dibasic amino acids cystine, ornithine, lysine and arginine (‘COLA’). Only cystine excretion is clinically relevant as it readily precipitates and may lead to stone formation. Cystinuria is responsible for approximately 5-10% of childhood UL and it is often associated with the formation of bladder stones. Cystine crystals have a characteristic hexagonal shape on microscopy. At a urinary pH of 5-7, cystine is poorly soluble and more likely to precipitate into stones.
- Hypocitraturia – citrate is a natural inhibitor of calcium phosphate and calcium oxalate formation within the urinary tract. It binds with calcium to form a soluble complex that then reduces the amount of free calcium in the urine. The lower the free calcium concentration, the less calcium is available to bind with oxalate, which is a risk factor for stone formation. Hypocitraturia may be seen in around 10% of children with UL (3). Chronic acidosis may lead to hypocitraturia due to increased proximal tubular reabsorption of citrate.
- Disorders of purine metabolism (predispose to the formation of purine stones including urate stones):
- Hyperuricaemia/hyperuricosuria secondary to chemotherapy (tumour lysis syndrome) and myelodysplastic syndrome (rare in children).
- Lesch-Nyhan syndrome (HGPRT deficiency) – an autosomal recessive disorder associated with hyperuricaemia/hyperuricosuria, formation of urate stones, abnormal choreoathetoid movements, self-mutilating behaviours (e.g. tongue biting), developmental delay, and progressive renal impairment.
- Adenine phosphoribosyltransferase (APRT) deficiency – an autosomal recessive disorder associated with build-up of 2,8-dihydroxyadenine (rather than adenine) and the formation of stones which are similarly radiolucent and have identical chemical reactivity to urate stones.
- Hereditary xanthinuria – an autosomal recessive disorder due to xanthine oxidase (XO) deficiency. XO converts xanthine to urate so xanthine build-up occurs leading to the formation of xanthine stones and hypouricaemia/hypouricosuria.
- Renal hypouricaemia – a disorder due to loss of function variants in the genes encoding the renal urate transporters (e.g. SLC22A12 encoding URAT1, and SLC2A9 encoding GLUT9). These variants reduce tubular urate reabsorption and lead to hypouricaemia/hyperuricosuria.
- Hyperoxaluria:
- Endocrine disorders:
- Hypo- and hyperthyroidism
- Cushing syndrome – can cause hypercalciuria
- Adrenal insufficiency
- Other conditions:
- Medullary sponge kidney (tubular ectasia)
- Fat necrosis
- Acute cortical necrosis (e.g. preterm infants with prolonged ischaemic down-time at birth) – these patients may have evidence of cortical (rather than medullary) NC.